Sen oppstart Kongenital binyreplasti
Medfødt adrenal hyperplasi er en arvelig gruppe av sykdommer der et sentralt enzym mangler fra kroppen.
Genetiske defekter tilstede ved fødselen (medfødt) påvirker flere enzymer som trengs for å produsere viktige binyrebarkhormoner.
Nesten 95 prosent av CAH-tilfeller er forårsaket av mangel på enzymet 21-hydroksylase. Kroppen kan ikke gjøre tilstrekkelige mengder av to viktige adrenalsteroidhormoner-kortisol og aldosteron – når enzym 21-hydroksylase mangler eller fungerer på et lavt nivå. Dette kaster av den delikate balansen mellom hormoner, hindrer riktig syntese av aldosteron og kortisol, og adrenal cortex begynner å lage androgener (mannlige steroidhormoner), som fører til mannlige egenskaper hos kvinner. I denne klassiske form av CAH kan saltbalanse også bli drastisk endret, noe som fører til elektrolytforstyrrelser, dehydrering og hjerterytmeendringer.
Mens mange pasienter diagnostiseres kort tid etter fødselen, er det en type sykdom som utvikles senere i livet, vanligvis i ungdomsårene eller tidlig voksen alder – det kalles ikke-klassisk eller forsinket CAH. Disse menneskene mangler bare noen av enzymene som er nødvendige for kortisolproduksjon. Aldosteronproduksjonen er ikke påvirket, så denne sykdomsformen er mindre alvorlig enn den medfødte formen og viser symptomer som ofte forveksles med PCOS, for eksempel:
For tidlig utvikling av kjønnshår ➢ Uregelmessige menstruasjonsperioder
Hirsutisme (uønsket eller overflødig kroppshår )
- Alvorlig akne (på ansikt og / eller kropp)
- Fertilitetsproblemer hos 10 prosent til 15 prosent av unge kvinner med CAH
- Diagnose av CAH
- Medfødt adrenalhyperplasi overføres genetisk. Øst-europeiske jøder (Ashkenazi) har økt risiko for å ha denne sykdommen, sammenlignet med befolkningen generelt. Siden CAH er en autosomal recessiv sykdom, må begge foreldrene ha et defekt enzymegenskap for å overføre det til barnet. På grunn av den genetiske overføringen av tilstanden, er mange mennesker oppmerksomme på risikoen i familien og la legen vite om behovet for genetisk screening. Legen kan utføre urin og blodprøver for å lete etter unormale kortisolnivåer eller andre hormonelle nivåer. Økte androgennivåer kan også vurderes ved diagnose. En grundig familiehistorie og fysisk eksamen er også nødvendig for at legen skal gjøre en komplett diagnose.
- Behandlingsalternativer
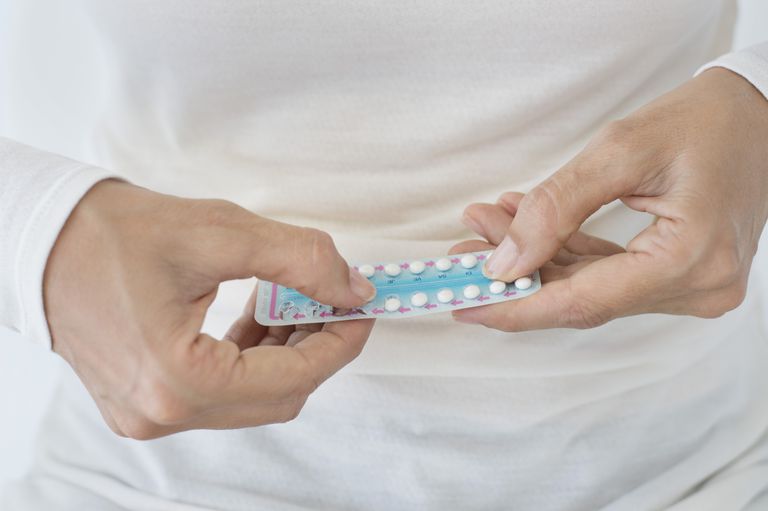
P-piller er vanligvis effektive for å regulere menstruasjonssyklusen, avtagende akne, og noen ganger unormalt hårtap. Hvis dette ikke er effektivt når det gjelder å håndtere symptomer, eller legen føler at p-piller ikke passer for deg, kan han eller hun vurdere å gi deg en lavdose-steroidbehandling.
Behandlingen er imidlertid ikke typisk livslang.
For personer med klassisk CAH med en aldosteronmangel, vil et stoff som fludrocortison (Florinef) beholde salt i kroppen. Spedbarn mottar også ekstra salt (som knuste tabletter eller løsninger), mens eldre pasienter med klassiske former for CAH spiser salt mat.